🎨Weblogo
- DNA, RNA, 및 단백질 서열의 로고 형태로 시퀀스 정렬을 생성하는 데 주로 사용되는 유명한 툴이다.
- 시퀀스 로고는 여러 시퀀스들의 정렬에서 특정 위치에서의 문자(뉴클레오티드나 아미노산)의 분포와 중요도를 시각적으로 표현하는 그림이다.
- 각 포지션마다 어떤 서열이 분포하는지
👉🏻 WebLogo의 특징
- 각 위치에서의 문자의 높이는 그 문자의 상대적 빈도나 중요도를 나타낸다.
- 로고의 높이는 각 위치에서의 정보 콘텐츠를 나타내고, 보통 비트(bit) 단위로 표현된다.
- WebLogo는 다양한 서열 정렬 형식, 예를 들어 FASTA, NBRF/PIR, ClustalW, GCG MSF, Phylip 등을 지원한다.
- 결과 로고는 PNG, SVG, PDF 같은 다양한 이미지 형식으로 출력할 수 있다.
- WebLogo를 활용하면 생물학자들은 서열의 모티프, 도메인, 그리고 다른 구조적 또는 기능적 영역의 일관성 및 변화를 쉽게 파악할 수 있다.
- WebLogo는 웹 인터페이스를 통해 온라인으로 사용할 수 있으며, 명령줄 도구로도 제공된다.
👉🏻 준비물
- 윈도우, 맥, 리눅스 등 운영체제
- 웹 브라우저 (크롬 등
- 파이썬 3
- 바이오 파이썬
👉🏻 순서
- 서열 다운로드
- 다운 받은 서열로 Multiple Sequence Alignment (MSA)를 수행
- MSA 결과로 weblog를 그리기
🎨 MSA
Multiple Sequence Alignment(이하 MSA)는 여러 개의 서열(주로 DNA, RNA, 단백질 서열)을 서로 정렬하여 서열 간의 유사성과 차이를 도출하는 방법이다.
MSA는 생물정보학 분야에서 아주 중요한 연구 도구로 사용된다.
간단한 예를 들자면, A, B, C라는 세 가지 생물의 유전자 서열이 있다고 가정하자.
각각의 유전자 서열은 서로 다를 수 있다.
MSA의 목적은 이 세 가지 서열을 최대한 잘 맞춰 정렬함으로써, 어느 부분이 유사하고 어느 부분이 다른지를 파악하는 것이다.
이렇게 MSA를 통해 얻어진 정보는 다음과 같은 연구에 유용하다:
- 진화적 관계 파악: 유사한 서열 구간은 공통의 조상으로부터 유래했을 가능성이 높다.
- 중요한 기능적 영역 식별: 여러 서열에서 보존된 구간은 그 부분이 중요한 기능을 담당할 가능성이 있다.
MSA는 다양한 알고리즘과 소프트웨어 도구를 통해 수행될 수 있다. 클러스터링, 다이나믹 프로그래밍 등의 방법들이 그 예이다.
1. NCBI에서 코로나 19 서열 받기
1-1. 검색창에 ncbi protein 검색
🎨 NCBI
NCBI는 "National Center for Biotechnology Information"의 줄임말이다.
미국의 국립 보건 연구소(NIH) 내에 위치한 기관으로, 생명 과학과 의학 분야의 정보를 수집, 보관, 배포하는 역할을 한다.
쉽게 말하면, NCBI는 생물학과 의학 연구자들을 위한 중요한 정보와 데이터베이스를 제공하는 웹사이트이다.
여기에는 유전자 서열, 단백질 구조, 논문 등 다양한 종류의 생물학적 정보가 저장되어 있다.
NCBI의 가장 유명한 서비스 중 하나는 "PubMed"로, 의학과 생물학 분야의 논문을 검색할 수 있는 데이터베이스이다. 또한 "GenBank"는 유전자 서열 정보를 보관하는 곳이다.
생물학과 의학 연구를 하지 않는 일반인에게는 NCBI가 직접적으로 필요하지 않을 수도 있지만, 생물정보학이나 관련 연구 분야에서는 NCBI의 리소스와 도구들이 매우 중요하게 활용된다.
https://www.ncbi.nlm.nih.gov/protein
Home - Protein - NCBI
www.ncbi.nlm.nih.gov
1-2. severe acute respiratory syndrome coronavirus 2 envelope protein
강사님은 covid envelope 치니까 자동완성으로 나오는데
나는 안 나와서 타이핑해서 검색
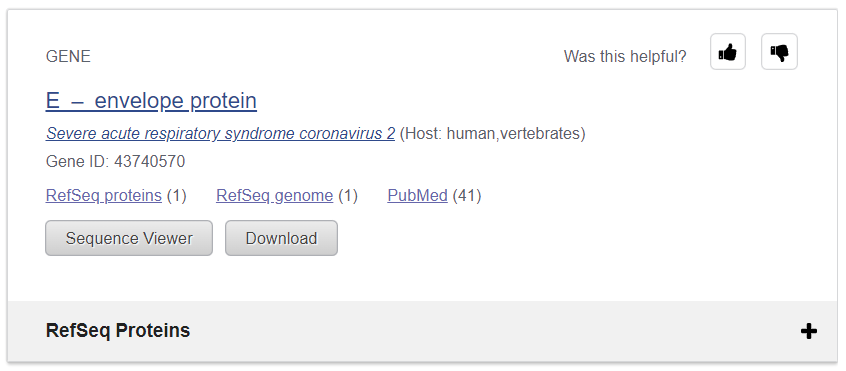

코로나 바이러스가 이렇게 여러가지 protein으로 구성됨
이 중에서 E에 해당하는 envelope protein을 받아 weblog를 그려보겠다
연구자들이 NCBI에 등록한 서열


3년만에 364만여개의 서열이 더 등록된 것..!
홈페이지에 보면 partial이라고 명시되어 있는 것이 있고 아닌 것이 있음

"Partial"은 영어로 "일부분의" 또는 "부분적인"이라는 뜻을 가진다.
코로나 바이러스 서열 관련 자료에서 "partial"이라는 표현을 본다면, 그것은 특정 서열이 완전한 전체 서열이 아닌 일부분만을 나타내는 것을 의미한다. 예를 들어, 코로나 바이러스의 전체 유전자 서열이 아닌, 특정 영역의 일부분만이 분석이나 샘플링 대상으로 포함되었을 때 "partial"이라는 표현을 사용할 수 있다.
간단히 말하자면, "partial" 서열은 해당 바이러스의 전체 유전자 서열 중 일부분만을 가리키는 것이다. 이는 전체 서열이 아닌 특정 부분만의 정보가 필요할 때나, 전체 서열을 얻기 어려운 상황에서 일부만을 분석하는 경우에 주로 사용된다.
이제 우리는 서열을 받아서 MSA 수행 전에 전처리로
Bio python을 가지고 partial이 아닌 것들만 가지고 진행을 하기 위한 필터링 작업 수행
다운 받기

send to 누르고

file 고르기

포맷은 FASTA
🎨 FASTA
FASTA는 생물 정보학에서 사용되는 서열 포맷 중 하나이다. 이 포맷은 DNA, RNA, 단백질 서열 등을 표현하기 위해 널리 사용된다. FASTA 포맷은 그 구조가 단순하여 인기가 있으며, 다양한 생물 정보학 도구와 프로그램에서 지원된다.
FASTA 파일의 기본 구조는 다음과 같다:
- 헤더 라인: '>' 문자로 시작하며, 이어서 서열의 이름 또는 설명이 온다.
- 서열 라인: 헤더 라인 다음에 오며, 실제 서열 데이터가 들어간다. A, T, C, G (DNA의 경우) 또는 단백질 아미노산 서열의 문자로 구성된다.
예를 들어 코로나 19 바이러스의 일부 서열이 FASTA 포맷으로 주어진다면:
>Sequence_Name_or_Description
ATGGAGAGCCTTGTCCCTGGTTTCAACGAGAAAACACACGTCCAACTCAGTTTGCCTGTTT
TACAGGTTCGCGACGTGCTCGTACGTGGCTTTGGAGACTCCGTGGAGGAGGTCTTATCAG위에서 'Sequence_Name_or_Description' 부분은 해당 서열의 이름이나 간략한 설명이 들어간다.
코로나 19의 경우, 유전자 서열 데이터를 다양한 데이터베이스에서 FASTA 포맷으로 다운로드 받을 수 있다. 이 파일을 사용하면 해당 바이러스의 유전자 구조를 분석하거나, 다른 서열과의 비교, 변이 점 검사 등 다양한 연구와 분석을 수행할 수 있다.

create file 누르기
파일명: covid.envelope.raw.fasta (전처리 하기 전이니까 raw로)로 저장
강사님은 867kb
나는 이미 mb 단위로 넘어갔다
3년만에 파일이 너무 커져서 다운 받는 데 오래 걸렸다
👉🏻 전처리
- amino acid 중 x가 있는 것은 제외를 한다
- amino acid: 아미노산, 단백질을 구성하는 기본 단위
- X는 모든 amino acid를 의미한다
- 더보기
아미노산은 생물학적으로 중요한 화학물질로, 단백질의 구성 요소이다. 생명체 내에서 단백질은 역할이 다양하며, 형태와 기능을 결정하는 중요한 역할을 한다. 아미노산은 마치 알파벳으로 이루어진 단어처럼 생물학적 언어의 문자로 생각할 수 있다.
아미노산은 기본적으로 세 가지 주요 부분으로 구성되어 있다:
- 아미노 그룹 (Amino Group): 이 부분은 질소 원자로 이루어져 있으며, 아미노산 이름의 "아미노"를 나타낸다. 이 부분은 모든 아미노산에서 공통으로 나타나며 질소를 함유하고 있다.
- 카르복실 그룹 (Carboxyl Group): 이 부분은 탄소, 산소, 그리고 수소 원자로 이루어져 있으며, 아미노산의 이름에 따라 "카르보실" 부분을 나타낸다. 이 부분은 모든 아미노산에서 공통으로 나타나며 아미노산을 구분짓는 중요한 특징 중 하나이다.
- 측기 (R-Group 또는 Side Chain): 이 부분은 각 아미노산마다 다르며, 아미노산 간의 차이를 만든다. 이 부분이 서로 다른 아미노산을 생성하고 단백질의 구조와 기능을 다양하게 만들어 준다.
아미노산은 단백질을 구성하는 단위로, 서로 다른 아미노산이 결합하여 다양한 단백질을 형성한다. 아미노산은 생명체에서 근육, 항체, 효소 등과 같은 중요한 생물학적 역할을 하며, 생명체 내에서 중요한 화학물질 중 하나이다.
- 각 서열을 unique하게 출력
- 중복 서열은 제거
- 같이 보는 방법도 있긴 함
- 최대한 포지션에 얼마나 다양하게 들어있는지 보기 위해
-
더보기
서열 분석에서 "description"은 서열 정보를 설명하는 부분을 가리키며, 서열 정보에는 DNA, RNA, 또는 단백질 서열과 관련된 추가적인 정보가 포함되어 있을 수 있으며, "description" 부분은 이러한 정보를 요약적으로 설명하는 부분이다.
"envelope protein"은 바이러스나 일부 다른 유기체의 단백질 중 하나로, 주로 바이러스 입자의 겉부분을 둘러싸고 있는 단백질을 가리킨다. 이러한 단백질은 바이러스 입자의 외피를 형성하거나 호스트 세포와 상호 작용하는 데 중요한 역할을 한다. "envelope protein"은 바이러스의 식별과 분류에도 사용된다.
서열 분석에서 "description"에서 "envelope protein"을 고른다는 것은 주어진 서열 데이터 중에서 "envelope protein"에 해당하는 부분만을 선택하거나 추출한다는 것을 의미하며, 이러한 서열 정보는 바이러스 연구나 다른 생물학적 연구 분야에서 중요한 정보를 제공한다.
🎮 확인해보기

- 파이썬에서 Biopython라이브러리의 SeqIO 모듈을 가져온다
🧩 Biopython
- 생물학적 데이터를 다루는 데 사용되는 파이썬 라이브러리
- DNA, RNA, 단백질 서열 등과 같은 생물학적 데이터를 다루고 분석하는 데 도움을 주는 다양한 도구와 함수 제공
🧩 SeqIO모듈
- Biopython에서 서열 데이터를 읽고 쓰는데 사용
- 주로 서열 파일을 읽어 파이썬 데이터 구조로 변환하거나 그 반대로 파이썬 데이터 구조를 서열 파일로 저장할 때 사용

- SeqIO 모듈을 사용하여 다운 받은 파일로부터 FASTA 형식의 서열 데이터를 읽어 파이썬에서 사용할 수 있는 데이터 구조로 파싱
- seq 변수에 FASTA 파일에서 읽어들인 서열 데이터가 저장
- SeqIO.parse(): Biopython 라이브러리에서 제공되는 함수, 서열 데이터를 파싱(해석 및 추출)하는 데 사용. 서열 데이터를 다양한 형식에서 읽고 파이썬 데이터 구조로 변환하는 데 도움을 준다.
- 기본 구문: SeqIO.parse(filename, format)
- foramt으로는 FASTA, GenBank, FASTQ 등의 서열 데이터 형식을 지정한다.
- 지정한 파일로부터 서열 데이터를 읽어들여 파이썬 데이터구조로 변환하고 반복 가능한(iterable) 객체로 반환

- 빈 집합(set)을 생성하고 이를 seq_set에 할당.
- 빈 집합을 초기화하고 나중에 집합에 원소를 추가하는 준비 단계


- FASTA 형식의 서열 데이터를 파일로 추출
- covid.envelope.fasta라는 파일을 생성하고 seq에 있는 각 서열 데이터를 순회하며 해당 서열을 화면에 출력한 후 스크립트 실행을 종료하는 역할
- 서열 데이터가 파일에 저장되거나 수정되지는 않는다.
- with open(): 파일을 열고 사용한 후 자동으로 닫아주는 구문
- 기본 구조: with open(filename, mode) as handle:
- mode: 파일을 열 때 모드, 읽기(r), 쓰기(w), 추가(a)등이 있다
- as handle: 파일을 handle 변수에 할당해 파일을 조작할 수 있다.
- for s in seq: seq 서열에서 데이터를 순회하며 각각의 서열을 s 변수에 저장한다
- print(s)를 통해 현재 순회중인 서열 s를 화면에 출력한다
- sys.exit(): sys 모듈을 사용해 현재 스크립트 실행을 즉시 종료
➡️ 결과

- 구성: ID, Name, Description, Seqeunce
- 여기서 Seq에 X가 있다면 제외
- Description에 Partial이 있다면 제외
🎮 제외하기

- 각 서열 데이터 내의 description에 partial 또는 truncated 라는 단어가 포함되어있는 경우 그 서열을 건너 뛴다

- 각 서열 데이터 내의 seq에 X가 있다면 그 서열을 건너뜀
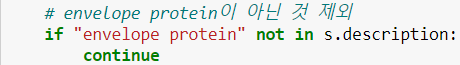
- 각 서열 데이터 내의 description에 'envelope protein'이 포함되어있지 않다면 그 서열을 건너뛴다
🎮 uniqe한 것들만 고르기

- 서열 데이터 중에서 중복된 서열을 제거하고 고유한 서열만을 파일에 기록
- 현재 순회중인 서열 s의 seq가 seq_set 집합에포함되어있지 않다면
- seq_set에 해당 데이터를 추가한다
- SeqIO.write(s, handle, "fasta"): 현재 서열 데이터 s를 파일에 기록
- FASTA 형식으로 현재 서열 s를 파일에 쓴다
- handle 은 앞서 with open()문을 통해 파일을 열었다
➡️ 결과

- 새 파일이 생성됨

- 3 줄이 하나의 레코드
👉🏻 Multiple Sequencing Alignment
- 다양한 서열이 주어진 경우, Multiple Sequence Alignment (다중 서열 정렬 또는 MSA)은 이러한 서열을 일렬로 정렬하여 각 위치의 서열 간 유사성을 파악하는 프로세스를 나타낸다. 이것은 생물학적 또는 유전학적 연구에서 매우 중요한 작업 중 하나이다
- MSA의 주요 목적은 서열 간의 유사성을 파악하여 공통된 특성을 찾아내고, 서열 간의 구조와 기능을 비교하는 것이다. 아래는 MSA를 설명하는 몇 가지 주요 개념이다.
- 서열 (Sequence): 서열은 DNA, RNA 또는 단백질과 같은 생물학적 분자의 일련의 문자 또는 염기로 이루어져 있으며, 특정 분자의 특징을 나타내며, 이들을 비교하고 연구하는 것이 중요하다.
- 다중 서열 (Multiple Sequences): MSA는 서열 그룹을 다룬다. 이 그룹은 종 또는 개체에 따른 여러 서열로 구성된다.
- 서열 정렬 (Sequence Alignment): MSA에서의 주요 작업은 서열을 서로 일치시키고 유사한 위치에 서열을 배치하는 것이다. 이렇게 하면 각 위치에서 서열 간의 차이와 유사성을 확인할 수 있다.
- 유사성 파악 (Detecting Similarity): 서열을 정렬하면 유사성을 쉽게 파악할 수 있다. 서열 간에 어떤 문자나 염기가 공통으로 나타날 때, 이것은 서열 간의 유사성을 나타낸다.
- 생물학적 의미 파악 (Biological Insights): MSA를 사용하면 공통된 기능, 구조 또는 진화적 연관성을 파악할 수 있다. 예를 들어, 단백질 서열 간의 MSA를 수행하면 특정 단백질 구조의 중요한 부분을 식별할 수 있다.
- MSA는 분자생물학, 진화생물학, 유전학 등 다양한 연구 분야에서 중요한 도구로 사용된다. 그것은 다양한 서열의 비교를 가능하게 하고 생물학적 의미를 발견하는 데 도움이 된다.
🧩 MUSCLE
https://www.ebi.ac.uk/Tools/msa/muscle/
MUSCLE < Multiple Sequence Alignment < EMBL-EBI
MUSCLE stands for MUltiple Sequence Comparison by Log- Expectation. MUSCLE is claimed to achieve both better average accuracy and better speed than ClustalW2 or T-Coffee, depending on the chosen options. Important note: This tool can align up to 500 sequen
www.ebi.ac.uk
"MUSCLE"은 다중 서열 정렬(Multiple Sequence Alignment)을 수행하기 위한 공개 소프트웨어 도구 중 하나이다. "MUSCLE"은 "Multiple Sequence Comparison by Log-Expectation"의 약자로, 서열 정렬을 위한 강력하고 효율적인 알고리즘을 제공한다. 이 도구는 서열 데이터를 일치시키고 서열 간의 유사성을 파악하여 다중 서열 정렬을 생성한다.
MUSCLE은 생물학자와 유전학자들 사이에서 매우 인기 있는 서열 정렬 도구 중 하나이며, 염기 서열 또는 아미노산 서열과 같은 다양한 유형의 서열 데이터에 대해 사용된다. MUSCLE을 사용하면 관련 서열 간의 공통 특성을 식별하고 진화적 관련성을 조사할 수 있다.
MUSCLE은 무료로 사용 가능하며, 명령줄 인터페이스나 다양한 생물정보학 소프트웨어 패키지와 통합하여 활용할 수 있다. 다양한 옵션과 매개변수를 통해 사용자가 원하는 정렬 설정을 조정할 수 있다.
- 웹에서 진행할 경우 제한사항

- 더 많은 서열을 하고 싶을 경우 다운 받아 로컬에서 수행해야 한다
🎮 파일 업로드



🎮 파라미터 설정
기본 옵션으로

🎮 제출

했는데

위에서 말했듯 강사님 파일은 2020년도 파일이라 데이터가 별로 없어서 바로 다음 단계로 넘어갔지만
내 파일은 3년치 데이터가 쌓인 큰 파일이라 다음으로 넘어갈 수 없었다.
🎮 데이터 자르기
50개 정도의 레코드만 새로운 파일로 만들어보려고 한다

- 빈 리스트에 레코드를 하나씩 추가한다
- 설정한 수 만큼 레코드가 추가되면 멈춘다
- 리스트에 저장된 레코드들을 새로 열린 fasta파일(output_handle)에 fasta형식으로 쓰는 작업을 수행한다.

- 새로 만들어진 파일로 다시 시도해보기



- 해당 부분 (+맨 윗줄 ~ 잘린 부분까지 전체 다) 드래그 해서 복사하기
🎮 weblogo
버클리에서 만든 웹로고 사이트
https://weblogo.berkeley.edu/logo.cgi
WebLogo - Create Sequence Logos
weblogo.berkeley.edu

- 복사한 내용 체크된 곳에 붙여넣기


- amino acid 체크
- 그냥 출력하면 한 줄로 쭉 나오기 때문에 Multiline 체크, 15개로 설정
- 나머지는 기본 설정
➡️ 결과

버튼 눌러주기

- 강사님 weblogo


- 대부분 E가 들어있고 K도 어느정도 들어 있다는 뜻

- 전부 다 S만 있다
- 알파벳과 알파벳의 크기는 아미노산 또는 염기 서열의 다양성과 중요성을 나타낸다
> 각 알파벳은 특정 위치에서 어떤 종류의 서열이 발견되는지를 나타낸다
- 여러가지 알파벳이 나타나는 경우, 해당 위치에서 서열 다양성이 높다는 것을 나타낸다.
- 일부 위치에서는 특정한 아미노산 또는 염기가 주로 나타나지만,
다른 위치에서는 여러가지 서열이 혼합되어 나타날 수 있다.
- 해당 위치에서 서열의 중요성과 다양성을 나타낸다
- 진화적 또는 생물학적으로 중요한 역할을 하는 위치일 수 있다.
> 한 번호에 두 가지 알파벳
- 해당 위치에서 다른 서열이 혼합되어 나타나는 것을 의미한다
- 예를 들어 S와 V가 함게 나타나는 경우 해당 위치에서 Serine(세린)과 Valine(발린)이 혼합된 서열이 나타난다는 것
> 알파벳의 높이는 해당 서열이 얼마나 빈도적으로 나타나는지를 나타낸다
- 해당 서열에서 얼마나 빈번하게 나타나는지를 나타낸다
- 더 큰 알파벳은 해당 서열이 더 자주 나타난다는 것을 의미
- 진화적으로 보다 보존되는 위치를 나타낼 수 있다.
> 알파벳 밑의 숫자
- 해당 서열의 위치
- 서열의 위치: 단백질 또는 DNA/RNA 서열에서 각각의 문자(아마노산 또는 염기)가 위치한 곳을 가리킨다
- 해당 서열의 문자열에서 각 문자의 순서를 의미
- 서열을 이루는 문자들이 나열된 순서를 나타낸다
출처: https://youtu.be/Nc4L4i7rlIg?si=lDd9HMxeQGltOi07
'데이터분석 > Bioinformatics' 카테고리의 다른 글
| [한주현님강의] DNA 분석 파이프라인(4) (0) | 2023.10.25 |
|---|---|
| [한주현님강의] DNA 분석 파이프라인(3) (2) | 2023.10.24 |
| [한주현님강의] DNA 분석 파이프라인(2) (0) | 2023.10.18 |
| [한주현님강의] DNA 분석 파이프라인(1) (1) | 2023.10.18 |
| [한주헌님강의] DNA 분석 파이프라인 - 소개편 (0) | 2023.10.17 |